About
I’m the Director of AI & Drug Design at Molecular Forecaster, Canada, where my focus is on leveraging machine learning, particularly deep learning, to address complex issues in chemistry and drug discovery.
My main research areas include:
- Developing strategies for molecular design using AI.
- Implementing explainable AI within the context of drug design.
- Applying deep learning for structure-based drug discovery methods.
Recent Works & Projects
Book Chapter : Toxicity Forecasts: Navigating Data-Driven AI/ML Models - From Theory to Practice
Submitted the first book chapter on “Toxicity Forecasts: Navigating Data-Driven AI/ML Models - From Theory to Practice” to be published by Taylor & Francis Group @AppleAcademicPress. Thank you Antoine Moitessier and Nicolas Moitessier for your invaluable support and pleasure to collaborate with you on this. The book chapter focuses on practical implementation of AI/ML with toxicity datasets and includes coding snippets from #rdkit #deepchem, and scikit-learn accompanied by detailed explanations. Thank you, Editors - Dr. Shrikaant Kulkarni and Dr. Shashikant V. Bhandari for your invite and support. Apple_Academy_Press. 
Project : Potency is All You need: Discovery of HDAC8 inhibitors to be added
Project : Generative Models for Fragment-based drug design
In the third CACHE challenge, we focused on developing new antivirals against SARS-CoV-2 by targeting the Mac1 domain of NSP3. We adopted an RNN-LSTM driven approach for molecular design, utilizing trained models for fragment expansion rather than designing whole molecules. This was based on structural data of the NSP3 Mac1 domain and its known fragment binders, particularly targeting the ADP binding site’s sub-sites. Then, using Artificial intelligence (AI)-guided and knowledge-based fragment merging and expansion approaches, we generated novel molecules that would serve as templates to identify highly similar compounds in the Enamine REAL database that would be commercially available. We proposed 91 potential compounds, which are currently being tested. This work showcases our commitment to open science and engaging the scientific community in our research.(Chemrxiv ). 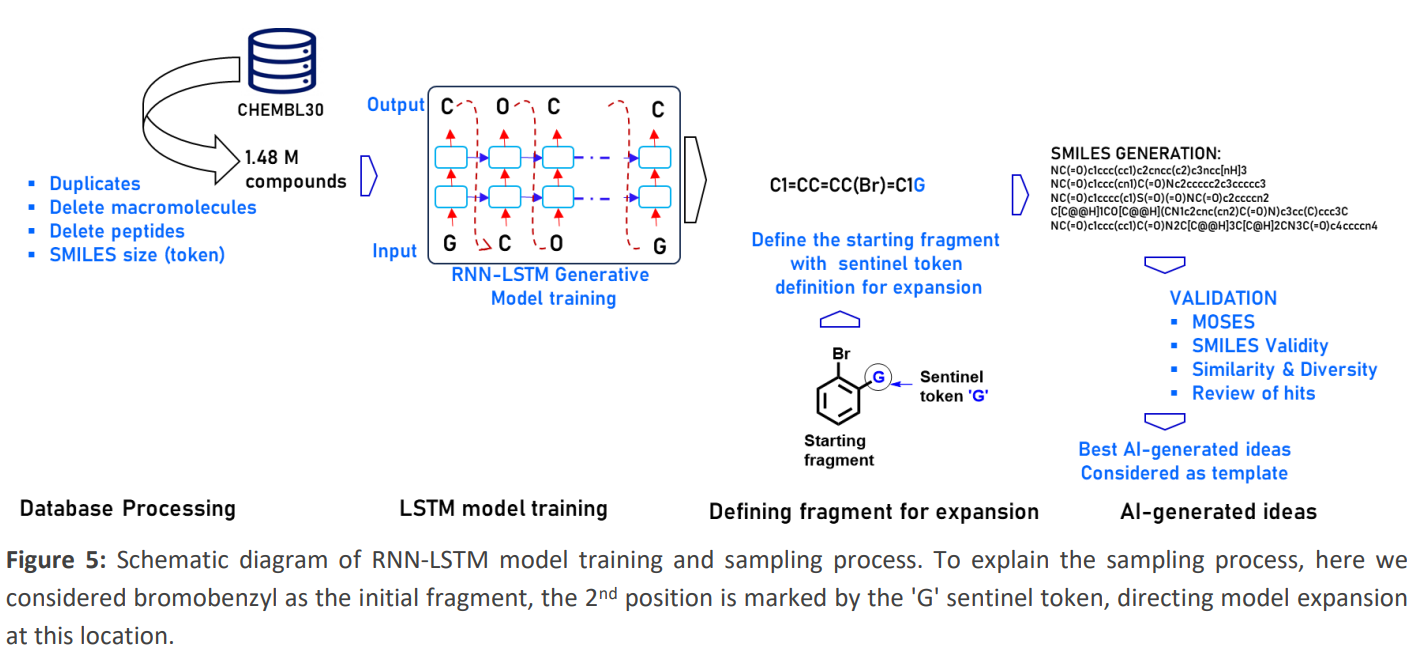
Project : Generative Models for Fragment-based drug design
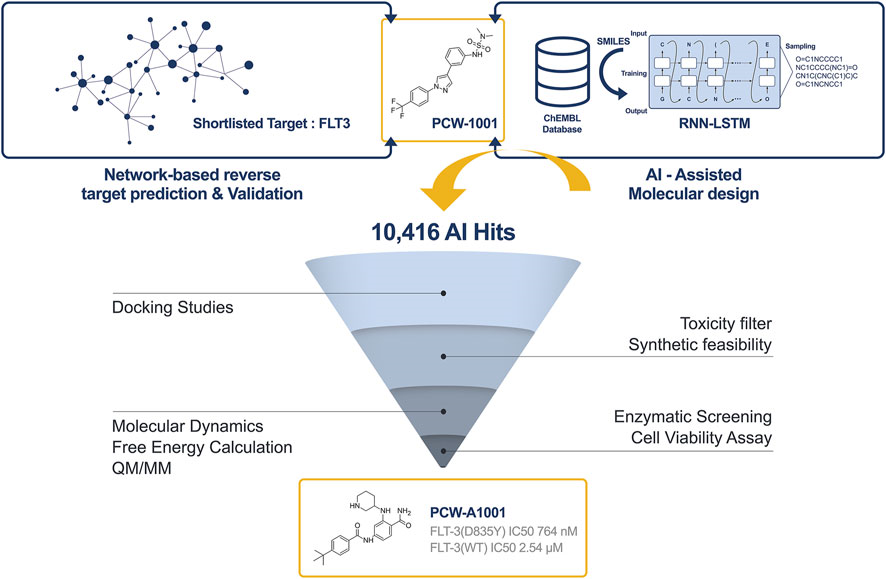
Treating acute myeloid leukemia (AML) by targeting FMS-like tyrosine kinase 3 (FLT-3) is considered an effective treatment strategy. By using AI-assisted hit optimization, we discovered a novel and highly selective compound with desired drug-like properties with which to target the FLT-3 (D835Y) mutant. In the current study, we applied an AI-assisted de novo design approach to identify a novel inhibitor of FLT-3 (D835Y). A recurrent neural network containing long short-term memory cells (LSTM) was implemented to generate potential candidates related to our in-house hit compound (PCW-1001). Approximately 10,416 hits were generated from 20 epochs, and the generated hits were further filtered using various toxicity and synthetic feasibility filters. Based on the docking and free energy ranking, the top compound was selected for synthesis and screening. Of these three compounds, PCW-A1001 proved to be highly selective for the FLT-3 (D835Y) mutant, with an IC50 of 764 nM, whereas the IC50 of FLT-3 WT was 2.54 μM. [Frontiers in Molecular Biosciences]